First step is to load seuratTools package and all other packages required
TLDR
seuratTools provides a single command to:
construct Seurat objects
filter genes by minimum expression and ubiquity
normalize and scale expression by any of several methods packaged in Seurat
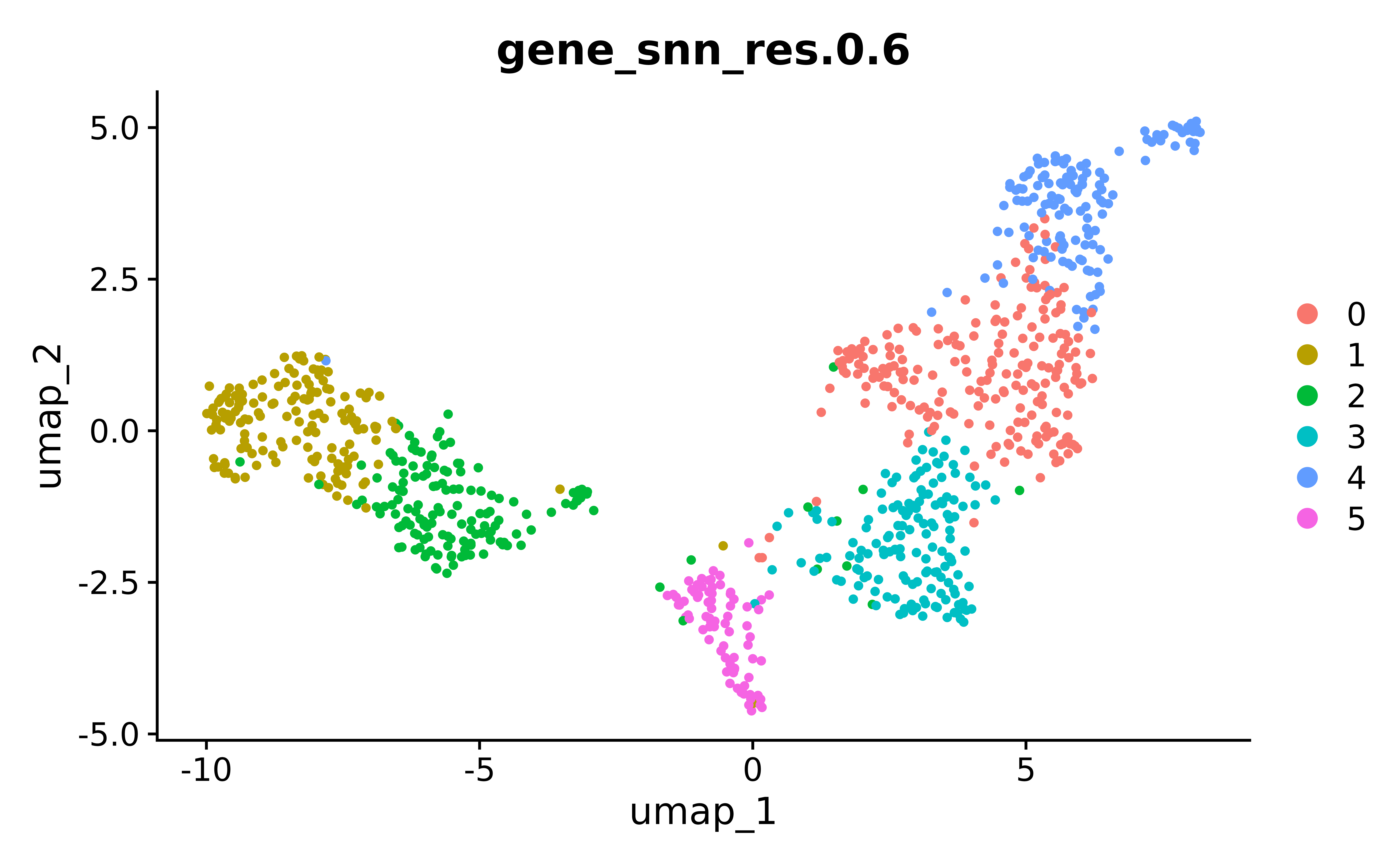
Run clustering on a single seurat object
By default clustering will be run at ten different resolutions between 0.2 and 2.0. Any resolution can be specified by providing the resolution argument as a numeric vector.
clustered_seu <- clustering_workflow(human_gene_transcript_seu,
experiment_name = "seurat_hu_trans",
organism = "human"
)
Get a first look at a processed dataset using an interactive shiny app
minimalSeuratApp(clustered_seu)Set up a seurat object
We start with a gene by cell matrix of count/UMI values
human_count[1:5, 1:5]| ds20181001-0001 | ds20181001-0002 | ds20181001-0003 | ds20181001-0004 | ds20181001-0005 | ds20181001-0006 | ds20181001-0007 | ds20181001-0008 | ds20181001-0009 | ds20181001-0010 | |
|---|---|---|---|---|---|---|---|---|---|---|
| 5-8S-rRNA | 0.00000 | 0.0000 | 0.0000 | 0.0000000 | 0.0000000 | 0.00000 | 0.0000000 | 0.0000 | 0.0000 | 5.557916 |
| A2M-AS1 | 0.00000 | 0.0000 | 0.0000 | 0.0000000 | 0.0000000 | 0.00000 | 0.0000000 | 56.8463 | 0.0000 | 0.000000 |
| A4GNT | 0.00000 | 0.0000 | 0.0000 | 0.0000000 | 0.0000000 | 0.00000 | 0.0000000 | 0.0000 | 0.0000 | 0.000000 |
| AADACL2-AS1 | 0.00000 | 0.0000 | 0.0000 | 0.0000000 | 0.0000000 | 0.00000 | 0.0000000 | 0.0000 | 0.0000 | 0.000000 |
| AAK1 | 67.89652 | 25.6852 | 182.6446 | 0.0627317 | 64.4965871 | 20.18323 | 0.2825439 | 659.7073 | 193.1156 | 0.000000 |
| AARS2 | 0.00000 | 0.0000 | 0.0000 | 0.0000000 | 63.2063514 | 0.00000 | 0.0000000 | 0.0000 | 0.0000 | 0.000000 |
| AATF | 130.11393 | 289.1958 | 349.4314 | 0.0000000 | 0.0000000 | 324.25505 | 126.6840636 | 0.0000 | 358.0968 | 0.000000 |
| ABBA01006766.1 | 0.00000 | 0.0000 | 0.0000 | 0.0000000 | 0.0000000 | 0.00000 | 0.0000000 | 0.0000 | 0.0000 | 0.000000 |
| ABCA10 | 0.00000 | 0.0000 | 0.0000 | 0.0000000 | 0.0000000 | 0.00000 | 0.0000000 | 0.0000 | 0.0000 | 0.000000 |
| ABCA4 | 84.15515 | 0.0000 | 0.0000 | 5.3283418 | 0.9109809 | 0.00000 | 58.8811160 | 0.0000 | 0.0000 | 180.928767 |
and a table of corresponding cell metadata
head(human_meta)| orig.ident | nCount_RNA | nFeature_RNA | sample_id | sample_id_1 | tissue_type | Kit_ID | Kit_sample | Seq_Number | Tissue.Type | Prep.Method | Prep.Number | Age | Time.Group | Poor_Read_Number | Moderate_Alignment | Rod_Cells | Possible_Rods | Non_Photoreceptors | Collection_Method | Outliers | VSX2_Outlier | X9_Cluster_Green_Rods | HR_Cluster_LB | HR_Cluster_Black | HR_Cluster_LG | HR_Cluster_DG | HR_Cluster_Pink | Cluster_Color | Fetal_Age | Old_Seq_Number | Old_Seq_Kit_ID | excluded_because | batch | names | type | gene_snn_res.0.2 | seurat_clusters | gene_snn_res.0.4 | gene_snn_res.0.6 | gene_snn_res.0.8 | gene_snn_res.1 | gene_snn_res.1.2 | gene_snn_res.1.4 | gene_snn_res.1.6 | gene_snn_res.1.8 | gene_snn_res.2 | read_count | percent.mt | nCount_gene | nFeature_gene | nCount_transcript | nFeature_transcript | transcript_snn_res.0.2 | transcript_snn_res.0.4 | transcript_snn_res.0.6 | transcript_snn_res.0.8 | transcript_snn_res.1 | transcript_snn_res.1.2 | transcript_snn_res.1.4 | transcript_snn_res.1.6 | transcript_snn_res.1.8 | transcript_snn_res.2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ds20181001-0001 | ds20181001 | 2384251.4 | 8908 | ds20181001-0001 | ds20181001-0001 | organoid | 1A | 1 | 3 | Organoid | Kuwahara | 115 | 56 | 1 | NA | NA | NA | NA | NA | FACS | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | keep | ds20181001_organoid | ds20181001-0001 | PE | 0 | 2 | 2 | 2 | 2 | 2 | 0 | 0 | 0 | 0 | 2 | NA | 0.1759367 | 526209.4 | 1532 | 525534.1 | 4290 | 0 | 2 | 1 | 1 | 1 | 0 | 1 | 0 | 0 | 0 |
| ds20181001-0002 | ds20181001 | 891702.8 | 6107 | ds20181001-0002 | ds20181001-0002 | organoid | 1A | 2 | 3 | Organoid | Kuwahara | 115 | 56 | 1 | NA | NA | NA | NA | NA | FACS | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | keep | ds20181001_organoid | ds20181001-0002 | PE | 0 | 2 | 2 | 2 | 2 | 2 | 0 | 0 | 0 | 0 | 2 | NA | 0.3751847 | 209036.3 | 1038 | 208914.3 | 2592 | 0 | 2 | 1 | 1 | 1 | 0 | 1 | 0 | 0 | 0 |
| ds20181001-0003 | ds20181001 | 1748316.1 | 9366 | ds20181001-0003 | ds20181001-0003 | organoid | 1A | 3 | 3 | Organoid | Kuwahara | 115 | 56 | 1 | NA | NA | NA | NA | NA | FACS | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | keep | ds20181001_organoid | ds20181001-0003 | PE | 3 | 12 | 5 | 5 | 5 | 6 | 7 | 5 | 7 | 4 | 12 | NA | 0.3964711 | 470723.7 | 1696 | 470707.7 | 4646 | 3 | 5 | 5 | 5 | 6 | 7 | 7 | 7 | 6 | 6 |
| ds20181001-0004 | ds20181001 | 2361597.0 | 8895 | ds20181001-0004 | ds20181001-0004 | organoid | 1A | 4 | 3 | Organoid | Kuwahara | 115 | 56 | 1 | NA | NA | NA | NA | NA | FACS | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | keep | ds20181001_organoid | ds20181001-0004 | PE | 3 | 12 | 5 | 5 | 5 | 6 | 7 | 5 | 7 | 4 | 12 | NA | 0.4962395 | 780500.9 | 1723 | 779109.3 | 4845 | 3 | 5 | 5 | 5 | 6 | 7 | 7 | 7 | 6 | 6 |
| ds20181001-0005 | ds20181001 | 1774650.6 | 7313 | ds20181001-0005 | ds20181001-0005 | organoid | 1A | 5 | 3 | Organoid | Kuwahara | 115 | 56 | 1 | NA | NA | NA | NA | NA | FACS | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | keep | ds20181001_organoid | ds20181001-0005 | PE | 0 | 2 | 2 | 2 | 2 | 2 | 0 | 0 | 0 | 0 | 2 | NA | 0.2448516 | 406661.3 | 1235 | 406722.9 | 3244 | 0 | 2 | 1 | 1 | 1 | 0 | 1 | 0 | 0 | 0 |
| ds20181001-0006 | ds20181001 | 1232401.4 | 6742 | ds20181001-0006 | ds20181001-0006 | organoid | 1A | 6 | 3 | Organoid | Kuwahara | 115 | 56 | 1 | NA | NA | NA | NA | NA | FACS | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | keep | ds20181001_organoid | ds20181001-0006 | PE | 0 | 2 | 2 | 2 | 2 | 2 | 0 | 0 | 0 | 0 | 2 | NA | 0.9387128 | 296053.0 | 1197 | 295994.0 | 2981 | 0 | 2 | 1 | 1 | 1 | 0 | 1 | 0 | 0 | 0 |
Then using these 2 datasets we can create a Seurat object in the
usual manner using the CreateSeuratObject function
myseu <- CreateSeuratObject(human_count, assay = "gene", meta.data = human_meta)
myseu
#> An object of class Seurat
#> 9740 features across 938 samples within 1 assay
#> Active assay: gene (9740 features, 0 variable features)
#> 1 layer present: countsPreprocess the seurat object
seuratTools includes a handy function to preprocess the data that handles normalization and scaling required for downstream analysis. Preprocessing is performed using existing Seurat functions. If needed, parameters can be specified by the user.
myseu <- seurat_preprocess(myseu)This single function includes sub-functions that normalizes, identifies highly variable features and scales the data:
- The normalizing sub-function performs log normalization using a default scale factor of 10,000.
preprocess_seu <- NormalizeData(myseu, verbose = FALSE)- After normalization, subset of features that exhibit high cell-to-cell variation in the dataset are identified. By default, 2,000 features per dataset are returned by this function.
preprocess_seu <- FindVariableFeatures(preprocess_seu,
selection.method = "vst",
verbose = FALSE
)- Finally, the data is scaled by applying linear transformation. This step shifts the gene expression, so that the mean expression across cells is 0 and scales the gene expression, so that the variance across cells is 1.
pre_process_seu <- ScaleData(preprocess_seu)Perform dimension reduction
seuratTools also implements a standardized dimension reduction step to select variable features at a user-specified threshold and perform PCA, tSNE, and UMAP. The default assay the dimension reduction is being run on is “gene”.
myseu <- seurat_reduce_dimensions(myseu, assay = "RNA")
DimPlot(myseu)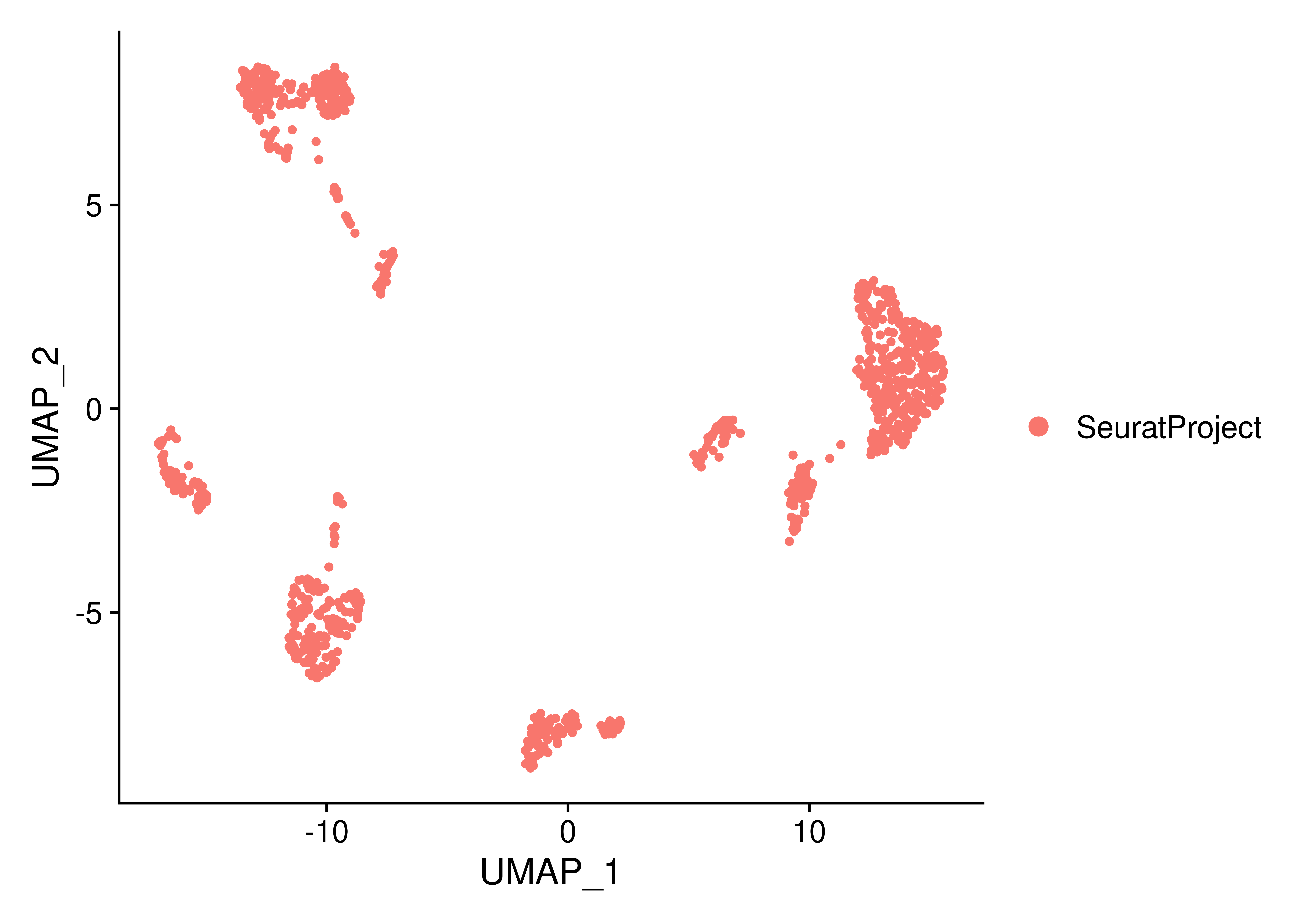
This function includes existing seurat functions which performs dimension reduction techniques.
- Perform PCA: Runs a PCA dimensionality reduction.
Dim_Red_seu <- RunPCA(myseu,
features = VariableFeatures(myseu),
do.print = FALSE
)- Perform UMAP: Runs the Uniform Manifold Approximation and Projection (UMAP) dimensional reduction technique.
Dim_Red_seu <- RunUMAP(Dim_Red_seu, dims = 1:30)Community detection by clustering
Clustering analysis is performed via Louvain(default) or alternative algorithms available in Seurat. Clustering is performed at a range of resolutions with default value ranging from 0.2 to 2 and pca reduction
seu <- seurat_cluster(seu = Dim_Red_seu, resolution = seq(0.2, 2, by = 0.2))This function produces clustering analysis via two steps performed using two different sub-functions
FindNeighbours: This function computes the nearest neighbors for a given dataset using k-nearest neighbor algorithm.FindClusters: The output from FindNeighbours is then used to identify clusters of cells based on clustering algorithm.